Introducing our new collaboration suite
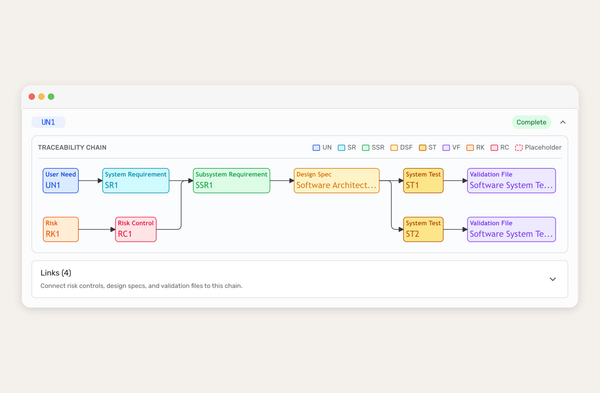
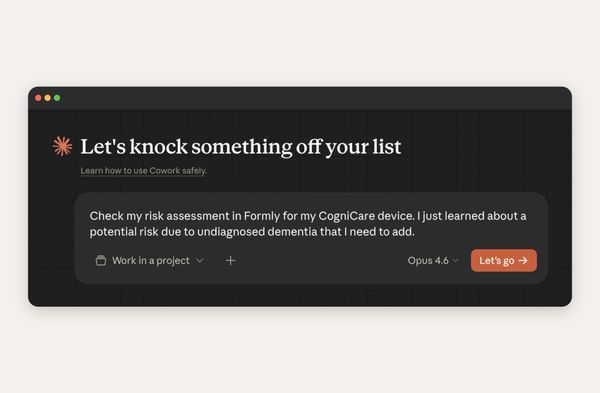
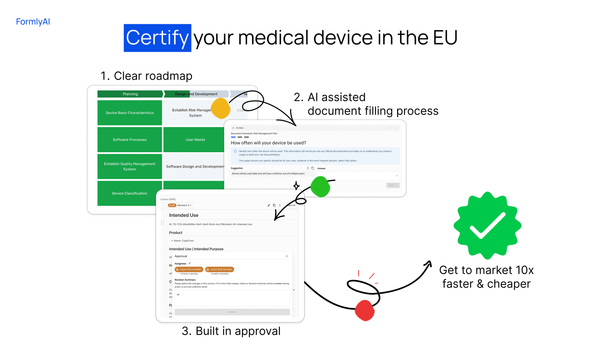
We built our own AI-native collaboration suite, and the conversation now includes the Dovetail agent. It reads every comment and redline in your document and acts on it, right inside your ISO 13485 quality system. A huge time saver for teams, and it's available now for all users.